

HIV-2 Tool Results
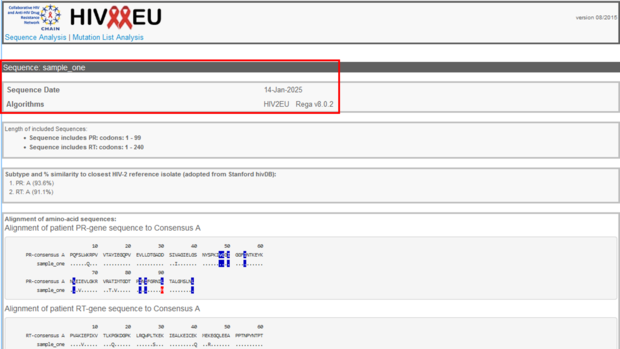
In the results view the top section provides information about the sequence name, the sequence date (which corresponds to the date of analysis on the HIV-GRADE website), and information about the algorithms used for the analysis.
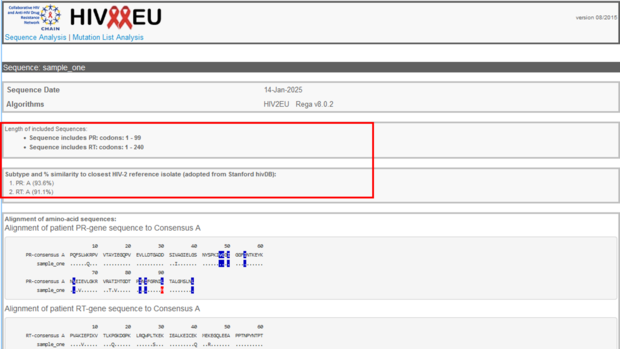
In the next section you will find information about the sequence itself, e.g. to which region of the HIV-2 genome the sequence belongs, which amino acid codons were analyzed and a prediction of the HIV-2 subtype and % of similarity to the closest reference sequence adopted from Los Alamos database. In our example, the submitted sequence corresponds to HIV-2 Protease, covers the amino acid codons 1-99, and Reverse Transkriptase (RT) covers aa 1-240. Sequence is predicted to be HIV-2 subtype A (93.6% in the Protease and 91.1% in RT).
If you have selected the “Mutation List Analysis”, the sequence information including the subtype will of course be omitted.
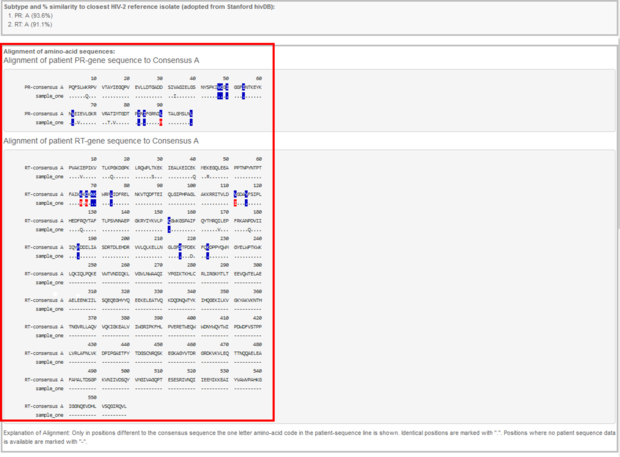
Below the subtype information you will find an alignment of the sequence to the subtype specific wildtype. All resistance relevant mutations are signed in blue, all detected resistance relevant mutations are signed in red.
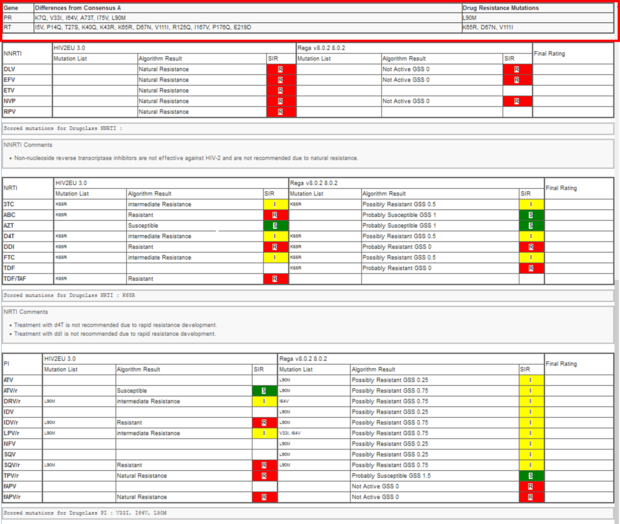
Next, you will find all amino acid differences of your submitted sequence compared to the consensus sequence and on the right hand the “Drug Resistance Mutations”.
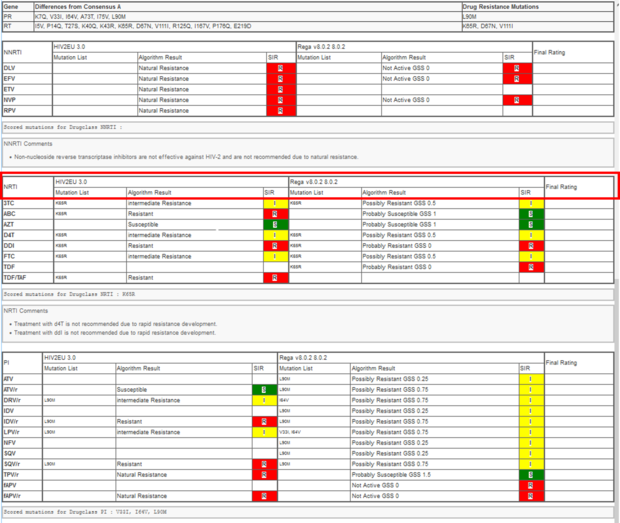
In the next section you will find a table with result from the algorithm used for interpretation of the different antiretroviral drugs sorted by drug classes. HIV-2 has a natural resistance to the NNRTI drug class. Therefore, all drugs here are resistant. The rows correspond to the different drugs (e.g. TDF/TAF (Tenofovir), DRV (Darunavir)) per drug class (NNRTIs, NRTIs, PIs, Integrase inhibitors IIs, entry inhibitors EI gp41) and the columns represent the result per algorithm, comprising mutation list, rating and SIR interpretation.
In our example the two algorithms have been used to analyze the submitted sequence (HIV2EU 3.0, and Rega v8.0.2 8.0.2).